多肽分子结构预测模块¶
多肽分子结构预测模块,基于精确的量子化学计算与完备的构象搜索法,获得肽分子系综,具有可靠完备的特点。能量范围广,包含10kacl/mol 范围内的完备构象。与其他方法相比,优势体现在:1.低能范围完备,理化性质计算准确。2. 高能区不遗漏,因此功能构象不缺失,更适合研究生化催化等问题。
使用方法¶
prothod init pepsearch
Prothod 输入配置文件 option.txt 和 peptide_list 输入文件
. # 输入的目录结构
├── option.txt
└── peptide_list
在 peptide_list 文件中写入要计算的氨基酸序列,如:

在 option.txt 文件中指定运行参数
#! The cookbook meta informations that you would cook (无需修改)
cookbook = 'pepsearch'
description = 'Batch perform structure search for peptide'
# 修改此行提交命令,ls_sub 后面跟的参数依次代表队列名,使用节点个数,每个节点使用核数。
run = 'ls_sub test 1 20 ./.default_script'
[option]
# 是否自动清除中间过程文件( true / false )
clean = true
执行 prothod run 即可即可提交计算。
运行结果¶
每个目录下面是计算获得的肽分子结构,例如
uni_xyz_list.txt 与 dihedral_angles_list2 两个文件分别存储每个结构的能量与二面角,例如
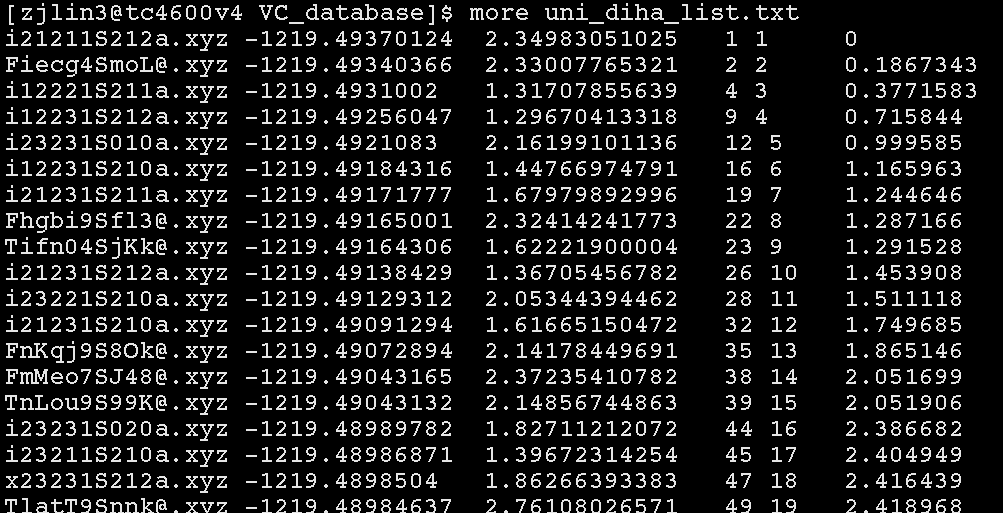
其中第一列是文件名,第二列是能量(原子单位),第三列是偶极矩,第五列是能量顺序,第七列是相对能量(偶极矩)
二面角文件
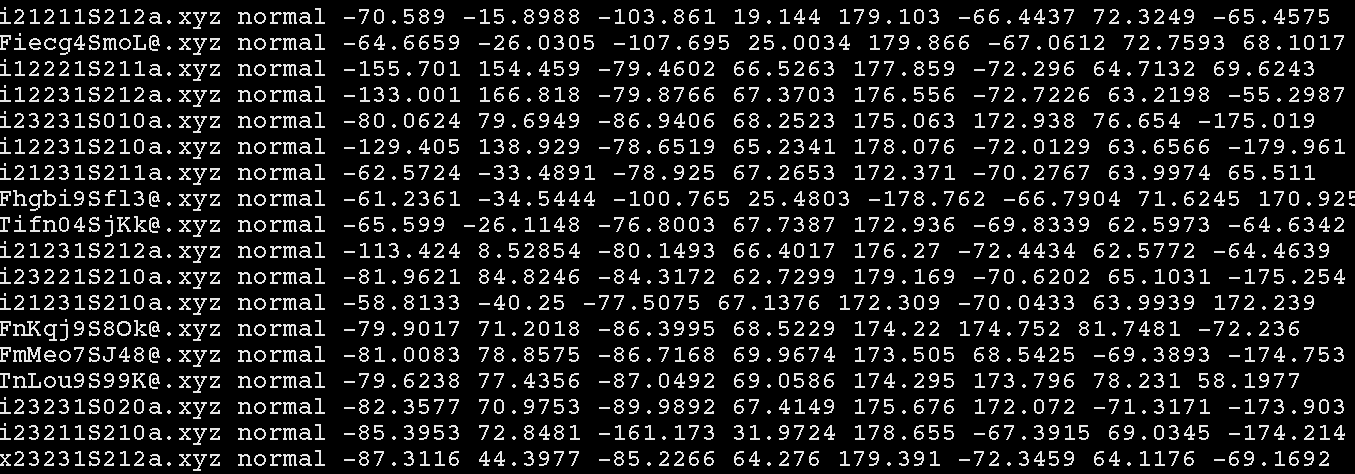
第一列文件名与uni_xyz_list.txt对应,从第三列开始是二面角,第1至2*N+1个二面角是主链二面角,其余为侧链二面角。