Getting started¶
多肽的结构预测的计算方法流程,可以抽象为采样,计算,筛选,等几个步骤。
整个流程中每一个步骤都可以使用不同的方法和不同的参数进行组合,而总体流程也可以进行变化。
我们在 Labkit 平台上面设计和实现了 molpack 包,可以方便的进行构象搜索的计算和方法探索工作。我们为每个步骤的常用操作提供了封装好的命令。
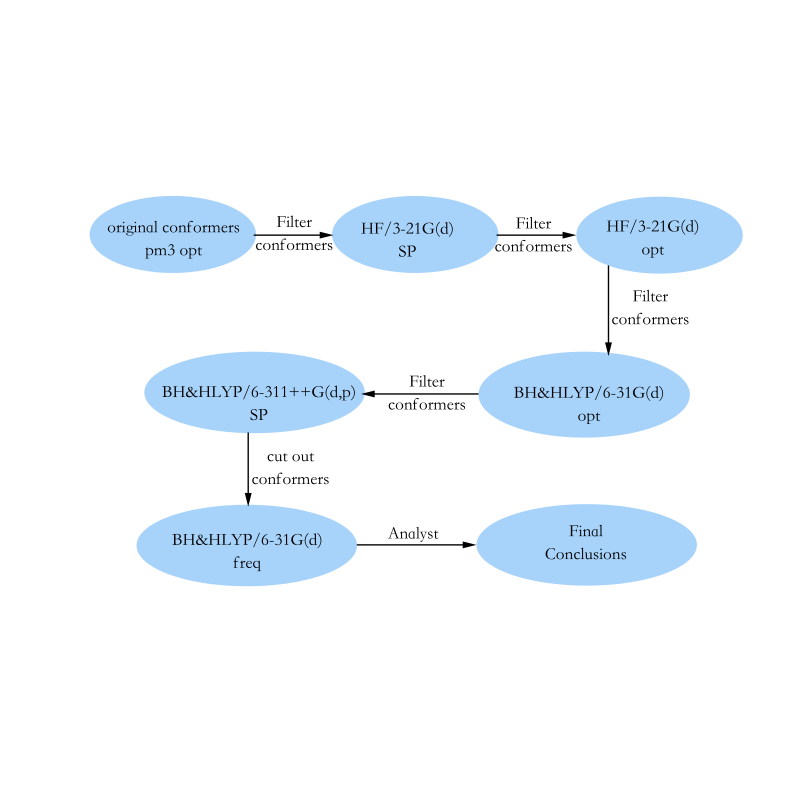
 计算流程¶
计算流程¶
势能面-》采样-》构型优化和计算能量,逐步简并筛选-》最终构型
拼接是一种采样方法。
molpack 模块¶
single 单步的计算操作,使用 sub 命令提交并行执行。
sample 采样操作,如构型生成,遗传算法等
conf 构型操作,例如构型的修饰,变形,拼接
tools 一些文件操作等小工具
ans 对数据进行非破坏性的分析,例如进行温度分布的分析
其他的在文件夹内的普通的操作,直接放在sh根目录里。
 示例¶
示例¶
m.single.gaussian, m.single.orca, m.single.dftb 调用 gaussian,orca,dftb 计算,规则是转换 xyz 文件为 out 文件。并且生成下一轮采用的 xyz 文件。
m.energy_cut [energy] 从当前目录中提取指定能量范围的结构。
findhf在当前目录下面对 out 文件以能量和偶极矩为标准进行构型的去重。拥有差别极小的能量和偶极矩的构型,视为相同构型。
ms.dihedral模块对于肽链,采用 biopython,根据设定的二面角定义,提取或设定二面角角度值序列。
da.sh 集合了处理程序可以直接得到温度分布。
 提交¶
提交¶
l.sub 是 Labkit 的队列功能的抽离独立版本,调用一个单独命令进行计算。适用于超算LSF 队列系统。
编写单体计算 single_file.sh 对数据进行 txt->out->findhf 管道方式处理,形成了固定接口的成熟的流程框架。
